2025年首篇!四川大学合作最新Nature
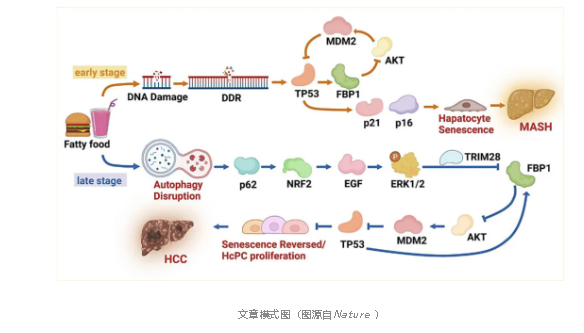
肝细胞癌(HCC)起源于分化的肝细胞在被病毒或代谢功能障碍相关的脂肪性肝炎(MASH)损伤的肝脏中进行代偿性增殖。在增加HCC风险的同时,MASH触发p53依赖性肝细胞衰老,这与营养过剩诱导的DNA断裂相似。这种肿瘤抑制反应是如何被绕过以允许致癌突变并使HCC进化的,这在以前是不清楚的。
FBP1首先在代谢应激的癌前疾病相关肝细胞和HCC祖细胞中下降,与AKT和NRF2的致瘤活性相似。通过加速FBP1和p53的降解,AKT和NRF2增强了先前衰老的HCC祖细胞的增殖和代谢活性。逆转衰老和支持增殖的NRF2-FBP1-AKT-p53代谢开关在小鼠和人类中起作用,也促进了DNA损伤诱导的体细胞突变的积累,这是MASH向HCC进展所必需的。

在促进MASH的同时,衰老可以抑制肿瘤的发生。尽管衰老样癌前肝细胞可能受到免疫监视,但MASH与免疫抑制和类似小鼠HCC祖细胞(HcPCs)的疾病相关肝细胞(daHeps)的积累有关。这些相反的结果提出了一个重要的问题,即MASH如何在引发复制性衰老的同时增加HCC风险。为了解决这个问题,研究人员确定了一个独特的、以FBP1为中心的信号传导和代谢开关,该开关可以逆转衰老并使MASH-HCC进展。
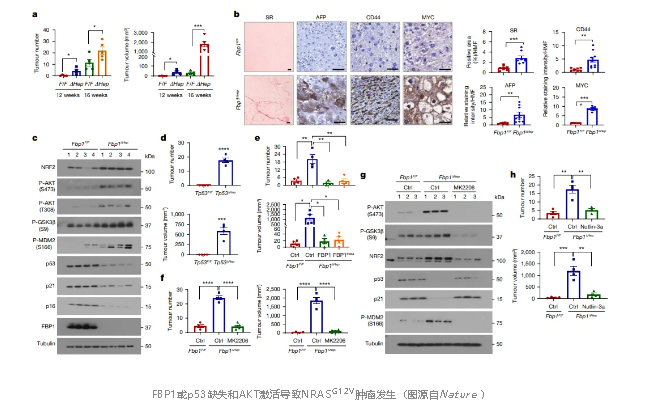
FBP1控制糖异生,抑制HCC。胚系FBP1缺乏合并葡萄糖剥夺可引发低血糖、乳酸性酸中毒、肝肿大、肝纤维化、肝损伤和高脂血症。尽管有饮食管理,FBP1缺乏的年轻人仍表现出MASLD的迹象。这是否会导致HCC尚不清楚。在FBP1缺陷小鼠中,研究人员发现饥饿诱导的低血糖是由糖异生损伤和糖原缺乏引起的,而MASLD体征是由AKT过度激活引起的。与酶活性无关,FBP1稳定地与AKT和醛缩酶B (ALDOB)结合,并招募蛋白磷酸酶2A催化亚基(PP2AC)来抑制AKT的活化。



EI Compendex,Scopus
2026年光电、材料、医工高新技术国际学术会议暨第三届人工智能、光电子学与光学技术国际研讨会(AIOT 2026)IOP出版
官方推荐




EI Compendex,Scopus,IEEE Xplore
2026年智能系统控制、优化与应用国际学术会议(ISCOA 2026)985主办
IEEE出版
